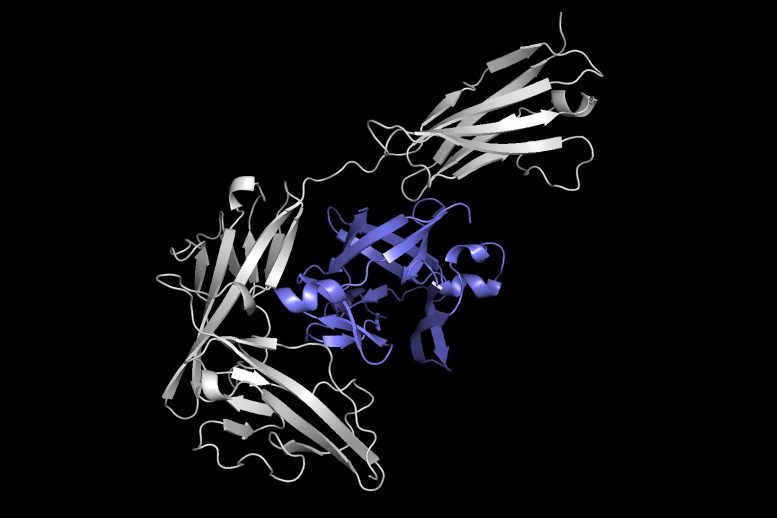
This picture exhibits one protein (in grey) docking with one other protein (in purple) to kind a protein complicated. Equidock, the machine studying system the researchers developed, can immediately predict a protein complicated like this in a matter of seconds. Credit score: Courtesy of the researchers
The machine-learning mannequin might assist scientists pace the event of recent medicines.
Antibodies, small proteins produced by the immune system, can connect to particular components of a virus to neutralize it. As scientists proceed to battle SARS-CoV-2, the virus that causes Covid-19, one attainable weapon is an artificial antibody that binds with the virus’ spike proteins to stop the virus from getting into a human cell.
To develop a profitable artificial antibody, researchers should perceive precisely how that attachment will occur. Proteins, with lumpy 3D constructions containing many folds, can stick collectively in thousands and thousands of combos, so discovering the precise protein complicated amongst virtually numerous candidates is extraordinarily time-consuming.
To streamline the method, MIT researchers created a machine-learning mannequin that may immediately predict the complicated that can kind when two proteins bind collectively. Their approach is between 80 and 500 instances sooner than state-of-the-art software program strategies, and infrequently predicts protein constructions which might be nearer to precise constructions which have been noticed experimentally.
This method might assist scientists higher perceive some organic processes that contain protein interactions, like DNA replication and restore; it might additionally pace up the method of growing new medicines.
“Deep studying is superb at capturing interactions between completely different proteins which might be in any other case tough for chemists or biologists to put in writing experimentally. A few of these interactions are very difficult, and other people haven’t discovered good methods to precise them. This deep-learning mannequin can study a majority of these interactions from knowledge,” says Octavian-Eugen Ganea, a postdoc within the MIT Laptop Science and Synthetic Intelligence Laboratory (CSAIL) and co-lead creator of the paper.
Ganea’s co-lead creator is Xinyuan Huang, a graduate pupil at ETH Zurich. MIT co-authors embrace Regina Barzilay, the Faculty of Engineering Distinguished Professor for AI and Well being in CSAIL, and Tommi Jaakkola, the Thomas Siebel Professor of Electrical Engineering in CSAIL and a member of the Institute for Information, Techniques, and Society. The analysis shall be introduced on the Worldwide Convention on Studying Representations.
Protein attachment
The mannequin the researchers developed, known as Equidock, focuses on inflexible physique docking — which happens when two proteins connect by rotating or translating in 3D house, however their shapes don’t squeeze or bend.
The mannequin takes the 3D constructions of two proteins and converts these constructions into 3D graphs that may be processed by the neural community. Proteins are shaped from chains of <div class="text-wrapper"><br />Amino acids are a set of organic compounds used to build proteins. There are about 500 naturally occurring known amino acids, though only 20 appear in the genetic code. Proteins consist of one or more chains of amino acids called polypeptides. The sequence of the amino acid chain causes the polypeptide to fold into a shape that is biologically active. The amino acid sequences of proteins are encoded in the genes. Nine proteinogenic amino acids are called "essential" for humans because they cannot be produced from other compounds by the human body and so must be taken in as food.<br /></div>"
</div></div>' data-gt-translate-attributes='["attribute":"data-cmtooltip", "format":"html"]'>amino acids, and every of these amino acids is represented by a node within the graph.
The researchers integrated geometric information into the mannequin, so it understands how objects can change if they're rotated or translated in 3D house. The mannequin additionally has mathematical information inbuilt that ensures the proteins at all times connect in the identical means, regardless of the place they exist in 3D house. That is how proteins dock within the human physique.
Utilizing this data, the machine-learning system identifies atoms of the 2 proteins which might be more than likely to work together and kind chemical reactions, often called binding-pocket factors. Then it makes use of these factors to position the 2 proteins collectively into a fancy.
“If we will perceive from the proteins which particular person components are more likely to be these binding pocket factors, then that can seize all the knowledge we have to place the 2 proteins collectively. Assuming we will discover these two units of factors, then we will simply learn the way to rotate and translate the proteins so one set matches the opposite set,” Ganea explains.
One of many greatest challenges of constructing this mannequin was overcoming the dearth of coaching knowledge. As a result of so little experimental 3D knowledge for proteins exist, it was particularly vital to include geometric information into Equidock, Ganea says. With out these geometric constraints, the mannequin may choose up false correlations within the dataset.
Seconds vs. hours
As soon as the mannequin was skilled, the researchers in contrast it to 4 software program strategies. Equidock is ready to predict the ultimate protein complicated after just one to 5 seconds. All of the baselines took for much longer, from between 10 minutes to an hour or extra.
In high quality measures, which calculate how intently the anticipated protein complicated matches the precise protein complicated, Equidock was usually comparable with the baselines, but it surely typically underperformed them.
“We're nonetheless lagging behind one of many baselines. Our methodology can nonetheless be improved, and it may well nonetheless be helpful. It could possibly be utilized in a really massive digital screening the place we wish to perceive how hundreds of proteins can work together and kind complexes. Our methodology could possibly be used to generate an preliminary set of candidates very quick, after which these could possibly be fine-tuned with among the extra correct, however slower, conventional strategies,” he says.
Along with utilizing this methodology with conventional fashions, the crew needs to include particular atomic interactions into Equidock so it may well make extra correct predictions. As an example, typically atoms in proteins will connect by way of hydrophobic interactions, which contain water molecules.
Their approach may be utilized to the event of small, drug-like molecules, Ganea says. These molecules bind with protein surfaces in particular methods, so quickly figuring out how that attachment happens might shorten the drug improvement timeline.
Sooner or later, they plan to boost Equidock so it may well make predictions for versatile protein docking. The most important hurdle there's a lack of information for coaching, so Ganea and his colleagues are working to generate artificial knowledge they might use to enhance the mannequin.
Reference: “Unbiased SE(3)-Equivariant Fashions for Finish-to-Finish Inflexible Protein Docking” by Octavian-Eugen Ganea, Xinyuan Huang, Charlotte Bunne, Yatao Bian, Regina Barzilay, Tommi S. Jaakkola and Andreas Krause, 28 September 2021, ICLR 2022 Convention.
OpenReview
This work was funded, partly, by the Machine Studying for Pharmaceutical Discovery and Synthesis consortium, the Swiss Nationwide Science Basis, the Abdul Latif Jameel Clinic for Machine Studying in Well being, the DTRA Discovery of Medical Countermeasures In opposition to New and Rising (DOMANE) threats program, and the DARPA Accelerated Molecular Discovery program.
Post a Comment